国家药监局关于发布晚期非小细胞肺癌临床试
1.新华制药「头孢拉定」首家过评
2.科济生物公布全人抗BCMA-CART临床结果
3.阿斯利康获「令泽舒」在华生产、商业化权利
4.上海医药拟2亿美元设立合资公司
5.药监局发布《晚期非小细胞肺癌临床试验终点技术指导原则》6.K药联合疗法新适应症获批新华制药「头孢拉定胶囊」首家通过一致性评价
9月17日,新华制药发布公告称,近日收到国家药监局核准签发的头孢拉定胶囊0.25g规格的《药品补充申请批件》,该产品通过仿制药一致性评价。新华制药成为国内首家通过头孢拉定胶囊(0.25g)仿制药一致性评价的企业。
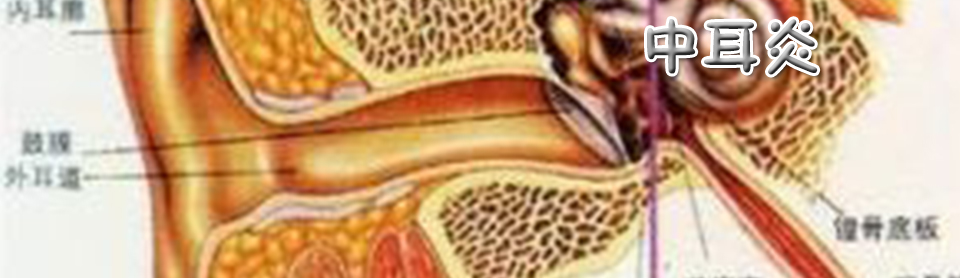
头孢拉定为美国BRISTOLMYERSSQUIBB公司研制成功的头孢菌素,属β-内酰胺类抗生素。适用于敏感菌所致的急性咽炎、扁桃体炎、中耳炎、支气管炎和肺炎等呼吸道感染、泌尿生殖道感染及皮肤软组织感染等。
年头孢拉定胶囊(商品名:泛捷复?)在中国本地化生产,持证商和生产厂家均为中美上海施贵宝制药有限公司。目前,于中国境内已上市的头孢拉定胶囊企业包括新华制药、齐鲁制药、湖南科伦制药等。
经查询相关资料,头孢拉定制剂年全球销售额约为万美元。截至年08月31日,新华制药在头孢拉定胶囊一致性评价项目上已投入研发费用约为人民币万元。
科济生物公布CT全人抗BCMA-CART细胞临床结果
近日,在第17届国际多发性骨髓瘤研讨会(InternationalMyelomaWorkshop,IMW)上,科济生物报告了CT全人抗BCMA-CART细胞治疗复发/难治多发性骨髓瘤的安全性和有效性研究的最新随访结果。
CTCAR-BCMAT是由科济生物自主研发的创新药物,是采用全人抗体靶向B细胞成熟抗原(BCMA)的嵌合抗原受体(CAR)修饰的T细胞,主要目标适应症为复发/难治多发性骨髓瘤。
上海交通大医院、医院和温州医院参加了该项探索性临床研究,医院血液科郝思国主任代表项目组在IMW会议上做了口头报告。截止年6月30日,共24例受试者接受CTBCMA-CART细胞输注,中位随访时间为天,总缓解率(ORR)为87.5%,完全缓解率(CR/sCR)为79.2%。
阿斯利康获「令泽舒」在华独家开发及商业化权利
9月18日,阿斯利康宣布已与Ironwood制药有限公司就双方关于令泽舒?(利那洛肽)的合作协议做了修订并达成共识。根据最新协议约定:阿斯利康获得利那洛肽在中国内地、中国香港和中国澳门的独家开发、生产和商业化权利。
利那洛肽是一种鸟苷酸环化酶-C激动剂,是美国消化病学会(AGA)指南推荐的一种治疗便秘型肠易激综合征(IBS-C)的创新药物,也是治疗IBS-C的标准药物。
中国Ⅲ期临床试验证实,与安慰剂组相比,利那洛肽可显著缓解IBS-C相关症状,其缓解程度反应者比例是安慰剂组的两倍以上(31.7%vs15.4%),疗效在治疗第一周就有体现,并在整个治疗期间症状都得到了改善。此外,由于利那洛肽作用于肠道局部,几乎不入血吸收,其安全性更为良好。
在年至年期间,阿斯利康将分三期向Ironwood支付总额为万美元的固定款项。此外,若能达成既定销售目标,Ironwood还能获得最高万美元的里程碑付款。
上海医药拟2亿美元设立合资公司
9月18日,上海医药发布公告,附属SPHPB与BIOCADHK正式签署《上海医药与BIOCAD间合资协议》与《关于SPH-BIOCAD(HK)Limited的股东协议》,将合资新设合资公司。合资公司作为BIOCAD在大中华区的唯一平台,未来将持续引入前沿生物医药产品和生物技术。
合资公司的注册资本金为4亿美元,其中上海医药以现金出资2.亿美元,占合资公司股权的50.1%;BIOCAD以现金2,万美元及6个生物医药产品在大中华区(包括中国大陆和港澳台地区)的永久、独家的研发、生产、销售及其他商业化权利作价出资,占合资公司股权的49.9%。
据了解,双方首批合作的6个生物医药产品包括3个生物类似药和3个创新生物药。3个生物类似药包括抗肿瘤药与类风湿性关节炎用药,皆已在俄罗斯上市销售,其中有2款还在俄罗斯以外多国获得批准上市。3个创新生物药分别为一款针对银屑病和强直性脊柱炎的IL-17抗体产品、一款PD-1抗体产品和一款GITR抗体产品,其中第1款产品已于今年在俄罗斯上市,第2款正处于申报审批阶段。
药监局发布《晚期非小细胞肺癌临床试验终点技术指导原则》
9月18日,为规范和指导我国治疗晚期非小细胞肺癌药物的临床试验设计和终点选择,提供可参考的技术规范,国家药品监督管理局组织制定了《晚期非小细胞肺癌临床试验终点技术指导原则》,现予发布。
Keytruda联合疗法新适应症获批,治疗子宫内膜癌
9月18日,默沙东(MSD)和卫材(Eisai)公司联合宣布,美国FDA批准默沙东的重磅PD-1抑制剂Keytruda,与口服酪氨酸激酶抑制剂Lenvima联用,治疗特定晚期子宫内膜癌患者。
这些患者不属于微卫星不稳定性高(MSI-H)或错配修复缺陷(dMMR)类型。她们在接受前期全身性疗法后疾病继续进展,且无法接受治愈性手术或放疗。这是Keytruda和Lenvima组合在美国首次获批。
值得注意的是,这一加速批准(acceleratedapproval)是在FDA实时肿瘤学审评(RTOR)试点项目下完成。而且依据FDA肿瘤卓越中心的另一试点项目Orbis,FDA,澳大利亚和加拿大的药品监管机构同时对这一申请进行了审评,今日同时在三个国家批准了这一申请。
这一批准是基于名为KEYNOTE-/Study的开放标签,单臂2期临床试验的结果。名转移性子宫内膜癌患者参加了试验。在名患者中,87%(94名)患者的肿瘤不属于MSI-H或dMMR。
在这94名患者中,Keytruda和Lenvima组合达到38.3%的客观缓解率(95%CI,29%-49%),完全缓解率为10.6%,部分缓解率为27.7%。在中位随访时间为18.7个月时,患者的中位缓解持续时间尚未达到。产生缓解的患者中69%生存期超过6个月。
▲以上新闻精选自Insight医药新闻Insight医药新闻,每天从多篇医药新闻中精选出4-8篇,只给你最重要的精选新闻实事。小新的话每天同一时间和你一起看药圈新闻Insight专利付费报告服务介绍Insight报告团队根据公开数据整理了FDA橙皮书中0~年以来上市药品的化合物专利信息,为企业提供不同类型的药品专利相关报告服务。
我们可以
提供FDA橙皮书中0~年以来上市药品的专利信息报告,展示药品专利美国到期时间和中国到期时间
结合FDA橙皮书中公布的NDA药品,提供同品种药品在国内注册申报、临床试验、上市信息等研发进展报告
转载请注明:http://www.zenmeshoutui.com/jbjc/6613.html